

The Cognitive Blind Spot
Why South Asian brain aging may start before APOE ever shows up

If a commercial Alzheimer’s risk report, longevity clinic, or genetic panel told you “you’re fine, no APOE ε4,” would that actually mean your brain is safe?
For a South Asian, the answer is almost certainly no.
Roughly 8.8 million Indians already have dementia, and the number is projected to climb steeply as the population ages in the LASI-DAD national estimate.
Globally, dementia cases are forecast to nearly triple, from 57 million in 2019 to roughly 153 million by 2050, with the smallest increases in high-income countries and the largest in the low- and middle-income regions that include most of South Asia in the GBD 2050 forecast. The same trajectory is already visible across the region, where dementia prevalence among older adults runs high, around 8% in Bangladesh, for example, in a national older-adult dementia study.
Yet APOE ε4, the most visible genetic risk marker in Alzheimer’s conversations, is present in only about 10% of South Asians. That is roughly half the European frequency (~15.5%) and a third of the African frequency (~26.8%) in a LASI-DAD APOE frequency analysis.
So if the gene most associated with Alzheimer’s is twice as rare in South Asians, why is dementia still rising so fast in our population?
It changes what we should screen for first.

The APOE ε4 Paradox
APOE ε4 is the strongest known genetic risk factor for late-onset Alzheimer’s in European populations. If you carry one copy, your risk roughly triples. Carry two, and you are looking at a ~12-fold increased risk in a large APOE risk synthesis.
The ε4 variant is the centrepiece of many genetic conversations about Alzheimer’s risk. Commercial reports and clinical panels built around European-derived data often make APOE the most visible genetic signal for Alzheimer’s prediction.

India’s largest dementia cohort, LASI-DAD, makes the paradox harder to ignore:
APOE ε4 frequency in Indians is ~10.8% in LASI-DAD. That means ~90% of South Asians would not be flagged by the most visible APOE ε4 signal on European-calibrated panels.
When ε4 IS present, it hits hard: in Indian cohorts, ε4 carriers have been reported to carry roughly 5.9 times higher odds of dementia (OR 5.90, 95% CI: 3.44–10.13) - a figure highlighted in the LASI-DAD deep-genomics analysis. The lever works. It is just pulled less often.
European-derived genetic risk scores barely work here: A study testing 56 known AD risk SNPs discovered through European GWAS found that genetic risk scores explained only 0.1–0.6% of variance in Indian episodic memory in a South Asian polygenic-risk replication study. The authors concluded that “genetic factors found predominantly through EA-GWAS may play a limited role in South Asians.”
South Asians have their own risk genes: The same 2026 LASI-DAD deep-genomics study screened 3.43 million rare and 100,618 common “Indian-enriched” genetic variants, variants more common in India than in European, East Asian, or African populations, for association with cognitive function. Implicated genes like ASAH1-AS1, DENND4A, and RAB11 do not appear on any European risk panel. Sex-specific effects were dramatic: one male-specific variant (PHF21A) was associated with a 10.66-point drop in HMSE score.
In plain language, South Asian Alzheimer’s risk does not map cleanly onto the European model.
If you screen a South Asian with a European-derived APOE panel and get a “normal” result, the test may still be missing the biology that matters most.
This is not just an amyloid story
This is where the South Asian pattern breaks from the Western Alzheimer’s playbook.
In European and North American clinical teaching, Alzheimer’s disease is amyloid-dominant. Vascular dementia is usually treated as the second major category, often estimated around 15–20% of clinically diagnosed dementia, with AD responsible for 60–70% (vascular dementia clinical review).
In South Asia, vascular cognitive impairment accounts for roughly 40% of the dementia burden according to an India-focused vascular cognitive impairment review. That is roughly double the Western estimate.
A northern Indian tertiary centre’s study of 1,876 dementia patients found the AD-to-vascular-dementia ratio was approximately 1.1:1 — nearly equal. When you add the 21% classified as “mixed dementia,” the combined vascular contribution approaches 50%.

The EDIS study from Singapore, comparing ethnic Indians, Malays, and Chinese, found that older Indians had a higher prevalence of cognitive impairment (~24.6%) than Chinese, and remained more likely to be cognitively impaired even after adjusting for cardiovascular risk factors in the population-based EDIS analysis. A companion EDIS neuroimaging study found Indians carried a higher burden of subcortical atrophy and distinct cerebrovascular and neurodegenerative imaging markers than Chinese in a Scientific Reports imaging paper.
And the BRAINS study - a landmark comparison of 1,126 Indian South Asians, 1,176 British South Asians, and 1,736 White British stroke patients - confirmed that South Asian stroke onset occurs 7 to 19 years earlier than in White British populations in the BRAINS stroke cohort comparison. And across South Asian stroke cohorts, the phenotype skews toward small-vessel disease rather than large-artery atherosclerosis.
The pattern is hard to miss: in many South Asian patients, brain aging is arriving through a microvascular pathway first. The conventional Alzheimer’s screen, built around amyloid plaques and APOE ε4, is tuned for a disease that often starts somewhere else.
Two Engines of Cognitive Decline in South Asians
With the data in front of us, I want to lay out what I think is the most clinically useful framing. South Asian cognitive decline is powered by two converging engines. Neither of them depends on APOE ε4.
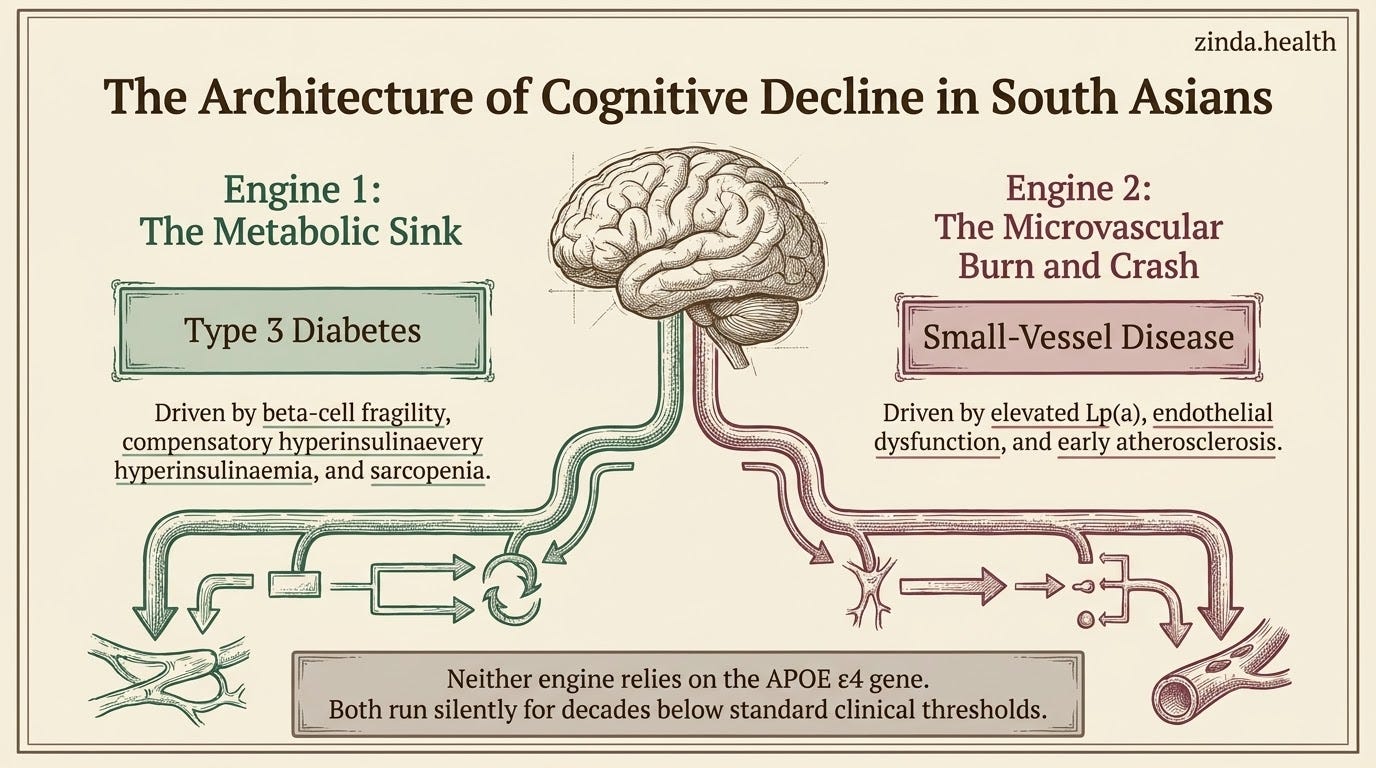
Engine One: The Metabolic Sink — “Type 3 Diabetes” And The IDE Competition
In 2005, Suzanne de la Monte and colleagues at Brown University published something that changed how neurologists think about Alzheimer’s in the original Type 3 Diabetes paper.
They showed that Alzheimer’s brains have extensive abnormalities in insulin, IGF-1, and IGF-2 signalling. Gene expression of these critical metabolic peptides and their receptors was reduced. The PI3K/Akt pathway, the backbone of insulin signalling, was impaired. The downstream consequences (increased GSK-3β activity, tau hyperphosphorylation, amyloid accumulation) looked like a metabolic disease, not a neurodegenerative one.
They called it Type 3 Diabetes: a brain-specific form of diabetes where insulin resistance and deficiency develop primarily inside the brain, overlapping with or occurring independently of the body-wide insulin resistance you measure with HbA1c.
This matters because it gives us a cleaner way to think about cognitive risk in South Asians.
One common South Asian branch is the insulin-resistant, compensatory-hyperinsulinaemic phenotype: lower BMI thresholds for metabolic syndrome, higher visceral adiposity at a given weight, sharper post-meal glucose spikes, and lower skeletal muscle reserves. But that is not the whole story.
The Zinda framework also assumes a Fragile Engine: beta-cells with lower reserve and earlier failure under load. Some patients therefore spend years in compensatory insulin excess, while others tip earlier into insulin-deficient phenotypes such as SIDD or Type 5 diabetes.
That distinction matters because the IDE mechanism applies most cleanly to the first branch: the insulin-degrading enzyme competition model.
Insulin-degrading enzyme (IDE) is a protease that your neurons rely on to clear amyloid-beta from the brain. IDE degrades both insulin and Aβ, using the same catalytic chamber. Crystal structures published in Nature in 2006 showed insulin and Aβ bound within the same IDE substrate-recognition system in the IDE crystal-structure study.
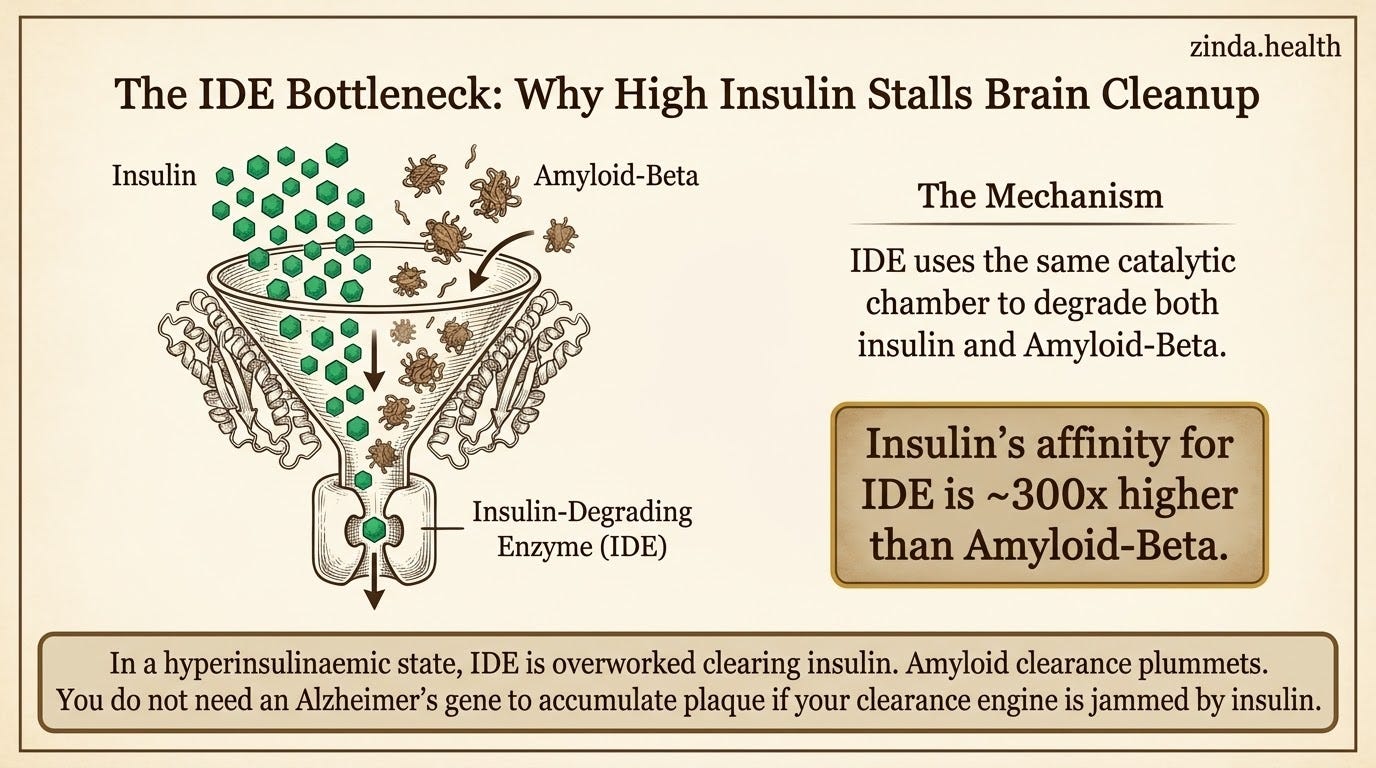
In a compensatory hyperinsulinaemic state, insulin can dominate that workload. The enzyme cannot distinguish between its two substrates. Insulin has an affinity for IDE (Km ~85 nM) that is roughly 300-fold higher than Aβ (Km ~25 μM). When insulin exposure stays high, IDE may spend more of its time clearing insulin, and Aβ clearance may suffer.
This is not theoretical. Farris and Tanzi showed in 2003 that knocking out the IDE gene in mice produced a >50% decrease in cerebral Aβ degradation, alongside hyperinsulinaemia and glucose intolerance in an IDE knockout study. Watson and colleagues then demonstrated in humans that experimental insulin infusion under euglycaemic conditions increased CSF Aβ42 levels, evidence that acute insulin exposure can modulate amyloid biology in vivo in a human insulin-infusion experiment.
The practical implication is narrower, but stronger: for South Asian patients who spend years in an insulin-resistant, compensatory-hyperinsulinaemic state, amyloid clearance may be impaired through the IDE bottleneck long before overt diabetes is diagnosed, regardless of APOE genotype.
Sarcopenia belongs in this picture too. South Asians carry some of the lowest skeletal muscle mass of any ethnic group. Muscle is the primary peripheral glucose sink. Mendelian randomization analyses support a causal relationship between low muscle mass / sarcopenia and cognitive impairment, with appendicular lean mass and walking pace causally linked to cognitive function in bidirectional MR. A Korean amyloid PET study found that higher thigh muscle mass was associated with significantly lesser risk of amyloid positivity (OR = 0.27 in females) in an amyloid PET body-composition study.
The chain is this for the insulin-resistant branch: low muscle mass → impaired glucose disposal → compensatory hyperinsulinaemia → IDE pressure → impaired amyloid handling → brain insulin resistance. It is one important route into the broader Type 3 Diabetes frame, not the only South Asian metabolic phenotype.
Engine Two: The Microvascular Burn And Crash
This is where Zinda’s framework maps directly to brain health.
We talk about the Burn and Crash across cardiovascular and metabolic domains. The same engine drives cerebral small vessel disease.
The SABRE study, which compared South Asian, European, and African Caribbean populations in the UK, found that diabetic South Asians had 63.3% more white matter hyperintensities (WMH) than diabetic Europeans in the SABRE brain MRI analysis. In the deep white matter regions, the excess was 102%. Each year of age led to an excess of 3.8% WMH load in frontal regions in South Asians compared to Europeans. And diastolic blood pressure predicted WMH in South Asians but not in Europeans — which means the vascular sensitivity of the South Asian brain to blood pressure is fundamentally different.
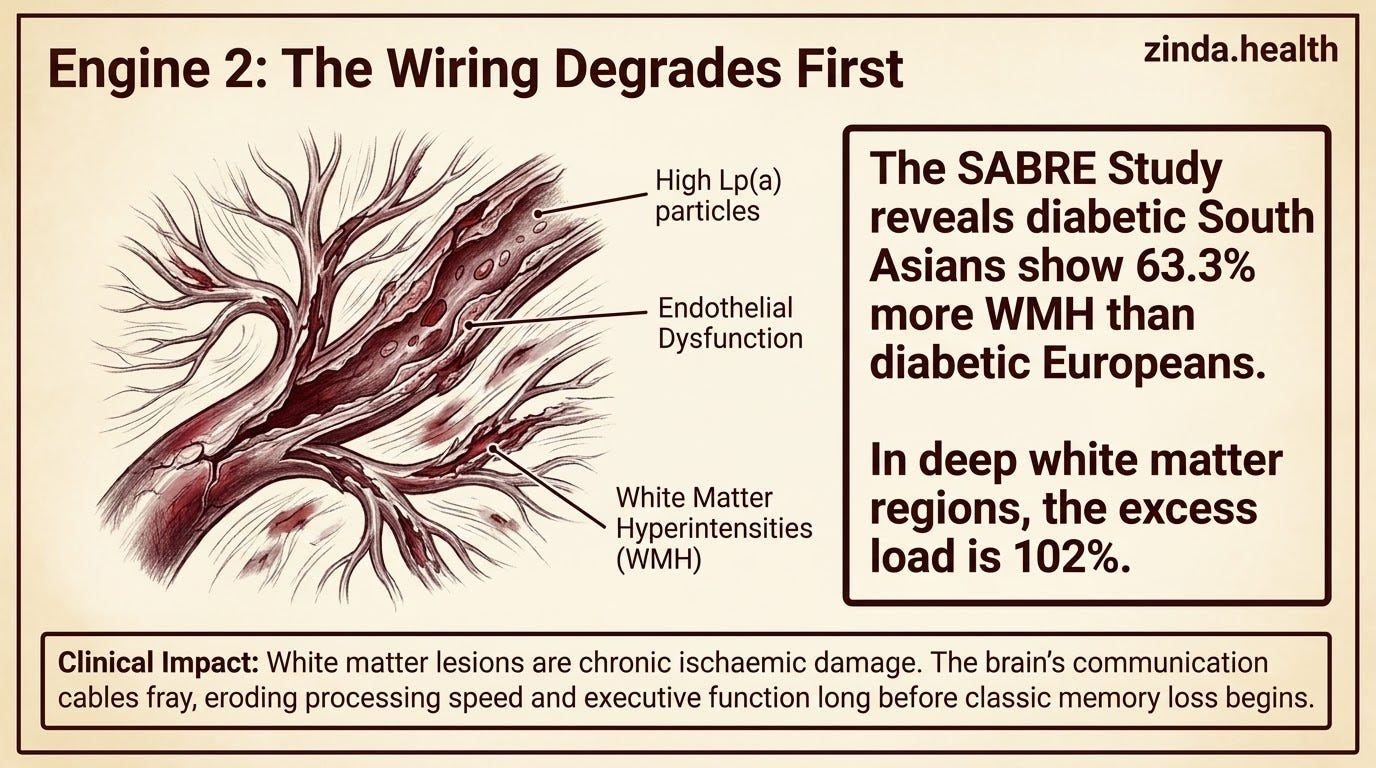
White matter hyperintensities are not incidental findings on an MRI.
Far too often we physicians treat these are chronic changes with no intervention to consider. That is a lazy practice of medicine. Reactive, not proactive.
These hyperintensities are evidence of chronic ischaemic damage to the brain’s wiring, the communication cables that connect one region to another. When your white matter is accumulating lesion load, your brain is losing its ability to route information efficiently. Processing speed drops. Executive function erodes. Memory retrieval slows. Eventually, the accumulated damage crosses a threshold where clinical dementia manifests, even in the absence of heavy amyloid burden.
Cerebrovascular reactivity, the ability of brain blood vessels to dilate in response to metabolic demand (measured via CO2 challenge testing), is emerging as an independent predictor of cognitive decline. A study in Neurology found that impaired CVR predicted cognitive performance independently of CSF Aβ42 and tau levels in a CVR biomarker study. CVR was a stronger predictor than white matter hyperintensity burden or vascular risk scores. A prospective study of adults with hypertension found that higher CVR in Alzheimer’s signature regions (precuneus, posterior cingulate, hippocampus) was associated with 40% lower risk of MCI in a hypertension CVR cohort.
For South Asians, who already carry higher Lp(a), more endothelial dysfunction, earlier atherosclerosis onset, and a propensity for small-vessel disease, the microvascular pathway is likely the primary engine of cognitive aging for many people who will never carry an APOE ε4 allele.
A lipidology nuance: lowering ApoB does not starve the brain
There is a common fear in healthspan/preventative medicine circles: “If I lower LDL too much, am I depriving my brain of cholesterol?”
This is the wrong model.
The brain is cholesterol-rich, but it largely runs its own cholesterol economy. Peripheral apoB-containing particles like LDL do not meaningfully deliver cholesterol into the brain because the blood-brain barrier separates plasma lipid traffic from central nervous system lipid traffic. Brain cholesterol is synthesized locally and trafficked through apoE-containing, HDL-like particles made largely by astrocytes and other glial cells. Adult neurons then acquire cholesterol through apoE-recognizing receptors such as LDLR and LRP1 (brain cholesterol homeostasis review; brain lipoprotein review).
Peter Attia went deep on this topic with Tom Dayspring this past week (the main reason this post was delayed).

That changes how to think about APOE4. APOE4 is not simply a “bad Alzheimer’s gene.” It is a different apoE protein shape in the brain’s lipid-transport system. If apoE4-containing brain lipoproteins traffic cholesterol less effectively, neuronal membrane cholesterol homeostasis can become unstable. And membrane cholesterol matters: experimental work shows that astrocyte-derived cholesterol can regulate neuronal amyloid-beta production by changing how APP interacts with secretases (PNAS cholesterol-amyloid study).
So the practical conclusion is almost the opposite of the folk fear. For a high-risk South Asian
patient, lowering ApoB and Lp(a)-linked vascular risk is brain-protective through the microvascular pathway, not brain-depleting. The safety literature is reassuring: a 2025 systematic review and meta-analysis of lipid-lowering trials found no significant association between lipid-lowering therapy and dementia or cognitive impairment, while confirming the need for better dementia-specific trials (lipid-lowering cognition meta-analysis).
The nuance is not “never worry about brain cholesterol.” It is: do not confuse plasma LDL-C with brain cholesterol supply. In select high-risk APOE4 patients, advanced clinicians may track emerging sterol markers such as desmosterol or 24S-hydroxycholesterol, but those are refinement tools — not a reason to leave ApoB high.
For the source conversation behind this framing, see Peter Attia’s episode with Tom Dayspring:
The screening stack: what Zinda would measure earlier
The conventional paradigm asks you to wait until you have memory complaints, then run a cognitive assessment and, when amyloid-targeting therapy or specialist risk stratification is on the table, discuss APOE ε4. For South Asians, that is too late. By the time Alzheimer’s disease manifests clinically, the underlying pathology has been accumulating for 15 to 20 years.